Understanding Vision Health
How The Eye Works
How The Eye Works
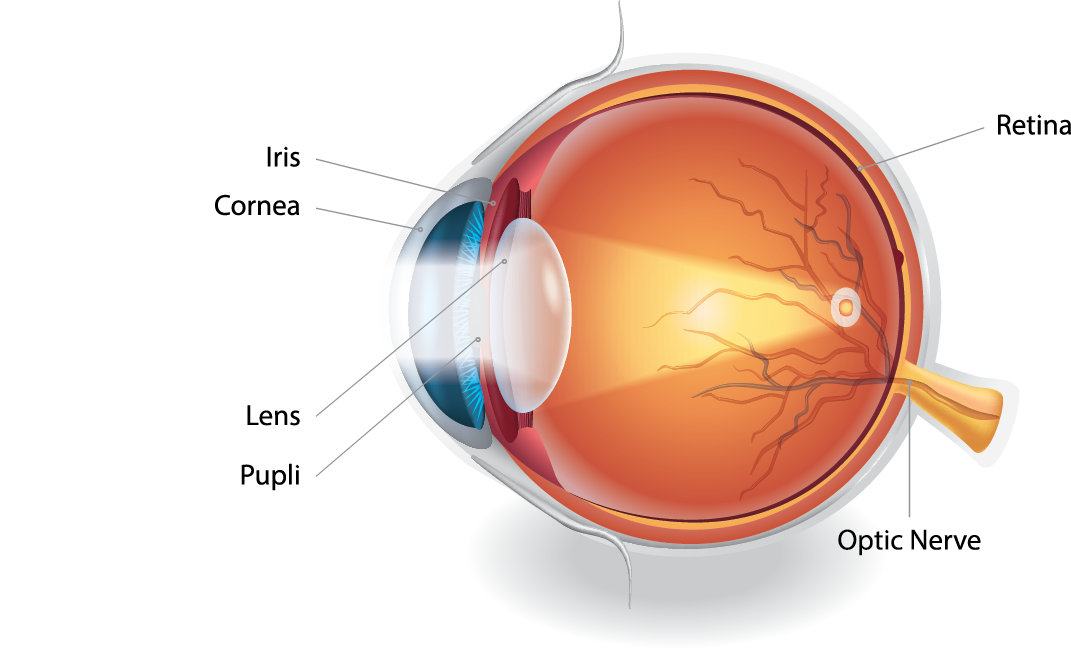
The retina sits at the back of the eye and plays a vital role in vision. It works a bit like a processor in a smartphone – taking in light and turning it into signals that the brain can understand.
When light enters the eye, the retina converts that light into electrical signals. These signals are then carried along the optic nerve to the brain, where they are interpreted as visual information.
The role of the retina in vision
The retina contains special light-sensitive cells called photoreceptors.
There are two main types:
- Rods: These help us see in low light and are responsible for night vision and side (peripheral) vision.
- Cones: These cells are mainly found in the central area of the retina and are responsible for detailed and colour vision.
What happens in retinal degeneration
In retinal degeneration (RD), the cells in the retina gradually stop working as they should. This means the signals sent to the brain can become incomplete or unclear, leading to changes in vision over time.
While rods and cones are most commonly affected, other retinal cells may also be involved, including:
- The retinal pigment epithelial layer
- Ganglion cells
- Bipolar cells within the neural layer of the retina
The exact pattern of vision loss can vary depending on the type of retinal condition and the underlying genetic cause.
Understanding how the eye works can help make sense of the different conditions that may affect vision.
Eye Conditions
Eye Conditions
There are many different eye and retinal conditions, and it can feel overwhelming to understand them all – especially after a new diagnosis.
Some conditions affect central vision, others side (peripheral) or night vision. Some are inherited, while others develop later in life. Each condition (and each person’s experience) is unique.
You do not need to understand everything at once. This section is here to help you find clear, reliable information when you are ready.
Eye and Retinal Conditions Supported by Retina South Africa
Retina South Africa provides information and support for people living with a wide range of eye and retinal conditions, including:
- Retinal disorders affecting the light-sensitive tissue at the back of the eye
- Age-related eye conditions, such as age-related macular degeneration (AMD)
- Diabetes-related eye conditions, including diabetic retinopathy
- Inherited retinal conditions, such as Retinitis Pigmentosa, Stargardt Disease, Usher Syndrome, Cone Rod Dystrophy, and Leber Congenital Amaurosis
- Rare retinal conditions that may affect colour vision, night vision, or retinal development
Eye Conditions Information Booklet
This downloadable PDF booklet provides clear, accessible information about a wide range of eye and retinal conditions.
It includes:
- An overview of how the eye and retina work
- Information on common and rare retinal disorders
- Age-related and diabetes-related eye conditions
- Inherited retinal conditions and genetic factors
- Current research, treatment approaches, and available support
Caring for Your Eyes
Caring for Your Eyes
There are many different eye and retinal conditions, and it can feel overwhelming to understand them all – especially after a new diagnosis.
Some conditions affect central vision, others side (peripheral) or night vision. Some are inherited, while others develop later in life. Each condition (and each person’s experience) is unique.
You do not need to understand everything at once. This section is here to help you find clear, reliable information when you are ready.
Become a Member
Become a part of our vibrant community to gain access to valuable support, insightful information, events, and the latest updates.
Everyday Steps to Support Eye Health
Good eye care includes:
- Having regular eye examinations
- Managing general health conditions such as diabetes and high blood pressure
- Protecting your eyes from sunlight and strain
- Maintaining a healthy lifestyle, including sleep, nutrition, exercise, and stress management
These steps are important for everyone, and especially for people living with retinal or inherited eye conditions.
Download the Eye Care Guide
This downloadable PDF guide explains practical, evidence-based steps you can take to support your eye health.
It includes:
- Regular eye tests and early detection
- Lifestyle factors such as smoking, sleep, diet, and exercise
- Managing blood sugar, blood pressure, and cholesterol
- Protecting your eyes from sunlight and excessive screen use
- Reliable sources of eye health information
Technology and Accessibility
Technology and Accessibility
Modern technology can play an important role in supporting independence for people with low vision. Many everyday devices now include built-in accessibility features that can be adjusted to suit individual needs.
For practical guidance, accessibility tips, apps, and training services, visit our Find Help & Services Page.